THE TARGETS


The main protease (3CL-PRO) is a key enzyme involved in viral replication and transcription. Hence, this is a very attractive antiviral target and several resolved structures have been already released. Some resolved structures are in complex with inhibitors thus allowing a precise characterization of its binding cavity. Similarly, screening studies proposed some potential 3CL-PRO inhibitors including both covalent and non-covalent ligands.

The papain-like protease of SARS-Cov-2 (PL-Pro) was recently resolved (PDB Id: 6W9C) in its apo form. A putative binding cavity was derived by the comparison with the corresponding structure from SARS-CoV co-crystallized with a potent inhibitor (PDB Id: 3E9S in complex with TTT).

The ADP ribose phosphatase nsp3 of SARS-Cov-2 was crystallized early this year (PDB Id: 6W02) in complex with the substrate ADP ribose, thus allowing a precise characterization of its catalytic cavity. Nsp3 is believed to play key roles in virus replication which goes beyond the simple phosphatase activity, although many of its functions remain to be investigated.

Nsp6 is a membrane spanning protein involved in the compartmentalized viral RNA synthesis. Until now, no crystal structure was resolved but a theoretical model can be generated by de novo modelling. Even though specific information concerning the nsp6 binding sites are not available, a recent analysis of known SARS-Cov-2 mutants highlighted the key role of a cluster composed of aromatic residues might be involved in the interaction with the membrane of the endoplasmic reticulum. Moreover, a potent nsp6 inhibitor (K22) was reported for several coronaviruses and its antiviral activity was ascribed to its capacity to interfere with the interaction between nsp6 and the membrane structure.

The nsp9 replicase protein of SARS-Cov-2 was recently resolved (PDB Id: 6W4B). This enzyme is believed to bind RNA-single strand in the viral replication complex. So far, no proper binding pockets have been identified on this protein. However, the crystal structure of nsp9 SARS-CoV (PDB Id: 1QZ8) suggests that molecules could bind close to the RNA binding site. In detail, the putative site for inhibitors can be derived by the observation that nsp9 is structurally homologous to the subdomains of serine proteases (PDB Id: 1P9U, in complex with the protease inhibitor PRD_000457) and to the first domain of human rhinovirus 3CLpros (PDB Id: 1CQQ, co-crystallized with RUPINTRIVIR a peptidomimetic inhibitor).

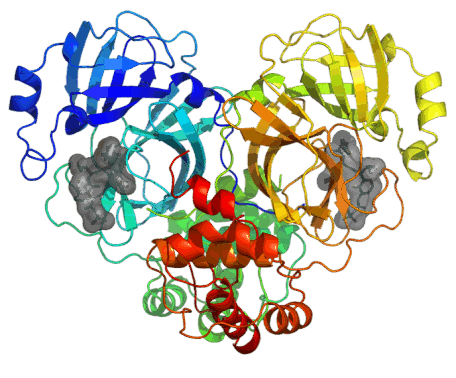
Nsp12 is an RNA-dependent RNA polymerase (RdRp) and is the main responsible for the RNA replication of the virus, making it an appealing target for the pharmacological treatment of the SARS-Cov-2 infection. Two structures of SARS-Cov-2 nsp12 in complex with its cofactors nsp7 and nsp8 were recently resolved (PDB Id: 6M71 and 7BV2). One of these structures (i.e. 7BV2) is co-crystallized with the inhibitor remdesivir, allowing the precise identification of the orthosteric site. Furthermore, a search for allosteric pockets was also performed since SARS-CoV nsp12 shows structural similarity with the RNA-dependent RNA polymerase of Hepatitis C virus (HCV), called NS5B as both enzymes share the well-known “hand” shape and NS5B is regulated by allosteric modulation.

The helicase nsp13 catalyzes the unwinding of oligonucleotides duplex into single strands. Given its vital role in virus replication, this has been pointed as a promising pharmacological target. For this reason, a homology model was built using the crystal structure of MERS-CoV nsp13 (PDB Id: 5wwp) as the template. The orthosteric site is highly conserved and a good agreement between the binding site residues of the crystal structure and the SARS-CoV-2 homology model was found. A putative allosteric site for nsp13 was also derived from the NS3 helicase of HCV (PDB Id: 4B75). A crystal structure of the SARS-CoV-2 Helicase was deposited very recently (PDB Id: 6ZSL).
HM 3D of structure of the SARS-CoV-2 Helicase NSP13 modelarchive

Nsp14 is a (guanine-N7) methyltransferase (N7-MTase) for mRNA capping. Currently, no resolved structure for SARS-Cov-2 nsp14 is available, but the protein can be modeled by homology techniques as the heterodimer nsp10-nasp14 using the available crystal structures of the SARS-CoV nsp10-nsp14 heterodimer (PDB Id: 5C8S) as the template. Luckily, the template was resolved in complex with the functional ligands involved in the catalytic reactions (namely the GpppA substrate and the demethylated SAH cofactor) thus allowing a precise characterization of the catalytic pocket within the modeled SARS-Cov-2 nsp14 structure. HM 3D structure of the SARS-CoV-2 NSP14-NSP10 hetero-oligomeric complex modelarchive

Nsp15 is a uridylate-specific endoribonuclease, the structure of which was recently resolved in complex with citrate (PDB Id: 6W01). The orthosteric cavity was derived by comparison with the corresponding resolved structures from SARS-CoV (PDB Id: 2H85). Even though both structures were in their apo form, the orthosteric cavity was identified based on sequence alignment and mutational analyses within the C-terminal domain. The so identified cavity was finally verified by preliminary docking simulations with the uridine 3′-phosphate ligand which produced a pose in encouraging agreement with that proposed for SARS- and MERS-Cov nsp15.

Nsp16 is a 2’O-methyltransferase and two resolved structures are available in complex (PDB Id: 6W4H and 6WKS) with its cofactor Nsp10 which is essential for the activity. The first structure is co-crystallized with SAM, while the second one includes both SAM and the P1-7-methylguanosine-P3-adenosine-5',5'-triphosphate (GTA) substrate thus allowing precise identification of the overall catalytic pocket.

The nucleocapsid protein (N-protein) of Coronaviruses is a structural protein, which packs the RNA genome forming a helical nucleocapsid structure or ribonucleoprotein (RNP) complex. Two X-ray structures of SARS-Cov-2 are already available (PDB Id: 6M3M and 6VYO) but in their apo form. However, five N-protein resolved structures from Human coronavirus OC43 (HCoV-OC43) co-crystallized with different ligands within the RNA-binding site (i.e., C5P, 5GP, U5P, AMP, P34 (inhibitor) with PDB Id: 4LMC, 4LM9, 4LM7, 4LI4, 4KXJ, respectively) are available. The binding site is extremely conserved, leading to easy identification of the corresponding pocket in the SARS-Cov-2 protein.

SARS-CoV-2 protein has attracted great interest in its role in the viral entry is the Spike protein, the receptor-binding domain (RBD) of which is recognized by the human receptor angiotensin-converting enzyme 2 (ACE2). Hence, different resolved proteins of the sole trimeric SARS-Cov-2 spike protein (PDB Id: 6LVN, 6LXT, 6VSB, 6VXX, 6VYB) as well as of its RBD in complex with ACE2 (PDB Id: 6LZG, 6M0J, 6M17, 6VW1) were recently reported. Rather than druggable pockets, these complexes allow a precise characterization of the protein-protein interactions by revealing the key regions involved in spike-ACE2 recognition. In the following docking simulations, the pocket analysis was performed by using mefloquine as the probe ligand since it was recently reported to be able to inhibit the spike-ACE2 interaction.

This scaffold-based library collects the most promising De Novo molecules as new potential pan Coronavirus inhibitors, selected as the top scored in the virtual screening campaign of 70 billion molecules against 19 SARS-CoV-2 Protein binding sites. Dompé has generated a huge virtual chemical space of hundreds of billions of compounds. This library was built starting from a database of millions of available commercial reagents that were combined using a set of robust synthetic reactions, in order to obtain a tangible chemical space, meaning that this is truly achievable in one reaction step. The reactions are encoded by means of a smart language that recounts detailed information about the reagent substructures directly involved in the reaction and their chemical environment, enabling an accurate annotation of each reaction in terms of synthetic feasibility.

The library contains drugs including the set of safe in man drugs, commercialized or under active development in clinical phases.

All the NATURAL COMPOUNDS molecular structures were selected from the FooDB server.

FOOD molecular structures coming from the FooDB server. FooDB is the world’s largest and most comprehensive resource on food constituents, chemistry and biology. It provides information on both macronutrients and micronutrients, including many of the constituents that give foods their flavor, color, taste, texture and aroma. FooDB server


The available peptides were generated by mixing in a combinatorial way all 20 natural amino acids to produce dipeptides, tripeptides, tetrapeptides and pentapeptides. All peptides have been constructed with an extended structure and have been optimized with MOPAC 2016. The protection consists in acetylation of the N-terminal end and the addition of amide in the C-terminal one.
Excalate4Cov
c/o Dompé Farmaceutici
Via Tommaso De Amicis, 95
80131 Napoli, Italy
Email: info@exscalate4cov.eu











